Neighbors¶
gpgraph-v2 defines two genotypes as neighbors when their distance is at most a cutoff. The default distance is Hamming; an alternative codon distance is available for codon-aligned DNA. Neighbor detection runs in Rust with rayon parallelism, with several specialized fast paths for common shapes.

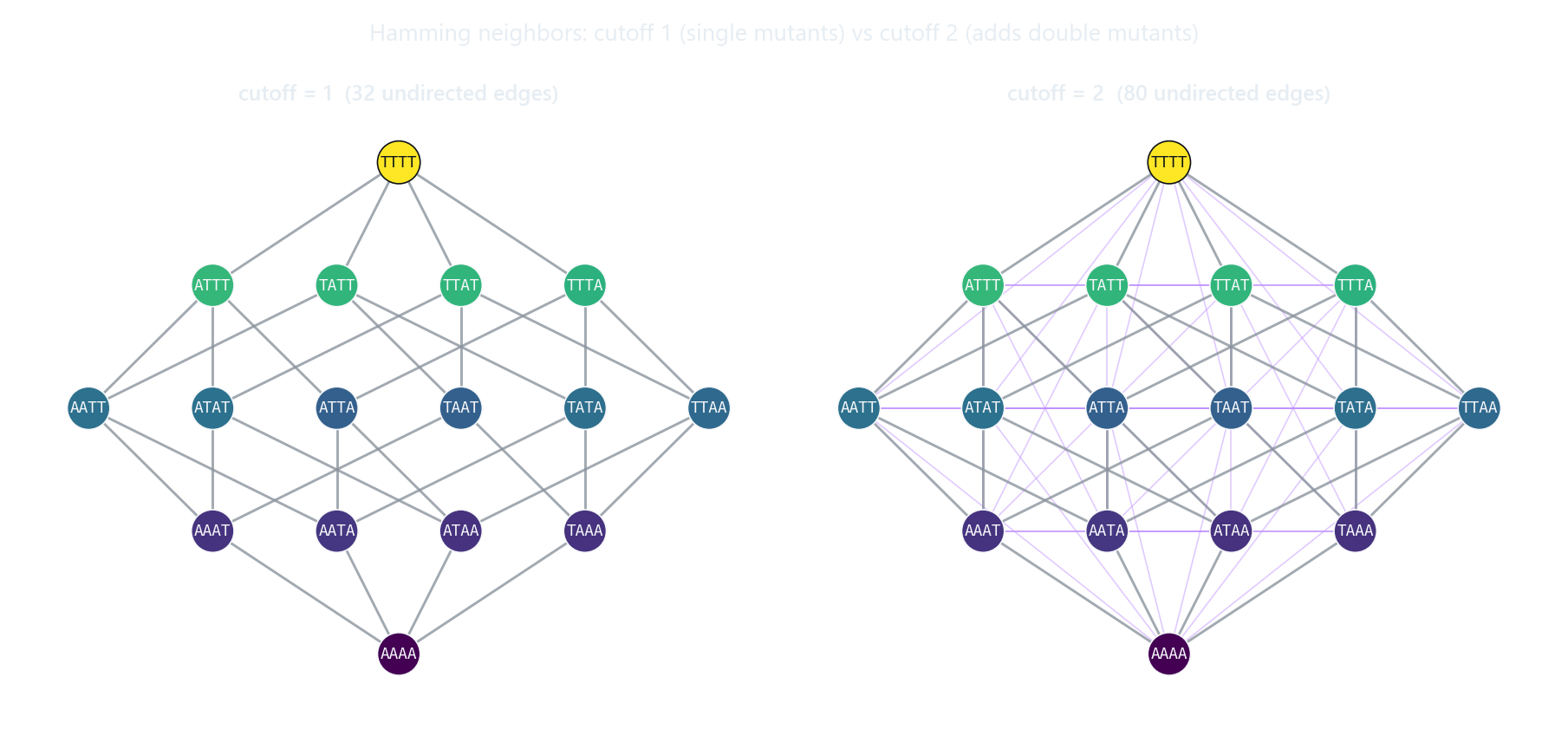
The cutoff sets how far apart two genotypes can be and still count as neighbors. Cutoff 1 gives the familiar single-mutation hypercube; raising it to 2 adds every double-mutant edge (the lighter diagonals above), which densifies the graph quickly.
Hamming distance¶
Hamming distance is the number of positions at which two equal-length sequences differ. Cutoff 1 means single-mutation neighbors; cutoff 2 includes double-mutants; and so on.
from gpgraph import GenotypePhenotypeGraph
G = GenotypePhenotypeGraph.from_gpm(gpm, neighbor_function="hamming", cutoff=1)
Codon distance¶
Codon distance is defined on length-3 codon segments: two codons are at distance d if their amino acids differ by at least d base-pair changes. Uses the standard genetic code.
The codon kernel expects the genotype length to be a multiple of 3 and the alphabet to be over {A, C, G, T}. Other layouts raise.
Custom distance functions¶
Pass any f(g1, g2, cutoff=...) -> bool callable:
def at_most_one_mismatch(g1: str, g2: str, cutoff: int = 1) -> bool:
return sum(a != b for a, b in zip(g1, g2)) <= cutoff
G = GenotypePhenotypeGraph.from_gpm(gpm, neighbor_function=at_most_one_mismatch)
The pure-Python O(N^2) fallback is used for user callables, so this is slow for large maps. Prefer one of the built-in kernels when you can.
Dispatch policy¶
gpgraph.neighbors.get_neighbors chooses the fastest available implementation based on the problem shape:
| Problem shape | Kernel | Complexity |
|---|---|---|
| User-supplied callable | Pure Python pairwise | O(N^2 * L) |
Hamming, biallelic binary_packed, cutoff <= 2 |
Rust bit-flip | O(N * C(L, cutoff)) |
Hamming, biallelic binary_packed, larger cutoff |
Rust rayon-parallel packed pairwise | O(N^2 * L) parallel |
| Hamming, non-binary alphabet | Rust rayon-parallel string pairwise | O(N^2 * L) parallel |
| Codon | Rust rayon-parallel codon pairwise | O(N^2 * L/3) parallel |
The bit-flip path at cutoff 1 or 2 is the dramatic win: for biallelic L=16 (n=65k), it is roughly 380x faster than a naive Python loop and 7x faster than the rayon-parallel pairwise variant.
The dispatch happens automatically inside from_gpm. You do not need to choose the kernel; the function inspects gpm.binary_packed to detect biallelic alphabets and picks the right entry point.
Edge representation¶
get_neighbors returns directed edges as a NumPy int64 array of shape (E, 2), sorted lexicographically:
import numpy as np
from gpgraph.neighbors import get_neighbors
edges = get_neighbors(gpm.genotypes.tolist(), neighbor_function="hamming", cutoff=1)
edges.shape # (E, 2)
edges.dtype # dtype('int64')
edges[:4]
# array([[0, 1],
# [0, 2],
# [0, 4],
# [1, 0]])
Each undirected pair appears twice, as (i, j) and (j, i). To convert to a Python list of tuples for nx.add_edges_from:
from gpgraph.neighbors import edges_array_to_tuples
edges_array_to_tuples(edges)
# [(0, 1), (0, 2), (0, 4), (1, 0), ...]
GenotypePhenotypeGraph.from_gpm does this conversion internally; you do not need to touch the array unless you are writing a custom graph builder.
Performance tip: pass binary_packed explicitly¶
If you are calling get_neighbors directly (not via from_gpm), pass binary_packed=gpm.binary_packed to trigger the bit-flip fast path:
from gpgraph.neighbors import get_neighbors
edges = get_neighbors(
gpm.genotypes.tolist(),
neighbor_function="hamming",
cutoff=1,
binary_packed=gpm.binary_packed,
)
Without binary_packed, the dispatch falls back to the string-comparison kernel, which is correct but ~3x slower.
Cutoff 0¶
cutoff=0 returns an empty edge array. Mostly a guard against off-by-one errors in callers.