What is a genotype-phenotype map?¶
A genotype-phenotype map (GPM) is the foundational data structure in epistasis-v2. It pairs each genetic sequence in your library with a measured phenotype value, for example a protein variant's fluorescence or binding affinity. All epistasis models in this library accept a GPM as their primary input, so understanding how to construct one correctly is the first step in any analysis.

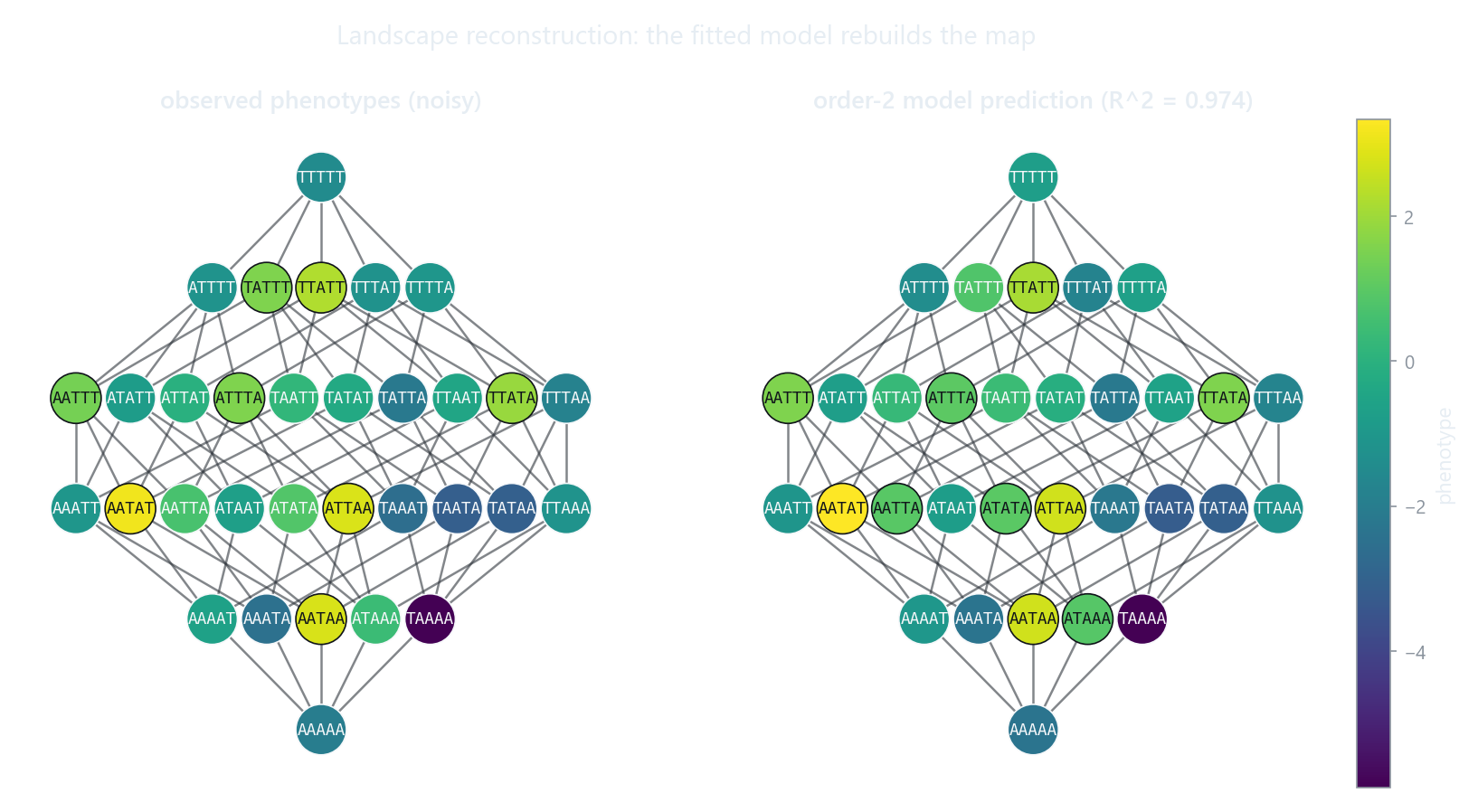
Each node is a genotype laid out by Hamming distance from wildtype and colored by phenotype. Fitting an epistasis model to the noisy observed map (left) recovers the smooth underlying landscape (right): the same GPM object is both the input you build and the output the model predicts.
The GenotypePhenotypeMap object¶
epistasis-v2 relies on gpmap-v2's GenotypePhenotypeMap class to represent your data. This object validates sequences, constructs a binary encoding of each genotype, and exposes the attributes that the epistasis kernels read internally.
Install gpmap-v2 alongside epistasis-v2:
gpmap-v2 is declared as a direct dependency and is installed automatically.
Constructing a GPM¶
Pass four arguments to GenotypePhenotypeMap:
| Parameter | Type | Description |
|---|---|---|
wildtype |
str |
The reference sequence. All other genotypes are measured relative to this sequence. |
genotypes |
list of str |
Every sequence in your library, including the wildtype. |
phenotypes |
list of float |
The measured phenotype for each genotype, in the same order as genotypes. |
mutations |
dict |
Maps each sequence position (0-indexed) to the list of allowed letters at that position. |
stdeviations |
list of float |
Optional measurement errors, one per genotype. Used by likelihood-based models. |
from gpmap import GenotypePhenotypeMap
gpm = GenotypePhenotypeMap(
wildtype="AAA",
genotypes=["AAA", "AAB", "ABA", "ABB", "BAA", "BAB", "BBA", "BBB"],
phenotypes=[0.1, 0.4, 0.3, 0.9, 0.2, 0.5, 0.7, 1.2],
mutations={0: ["A", "B"], 1: ["A", "B"], 2: ["A", "B"]},
stdeviations=[0.05, 0.05, 0.05, 0.05, 0.05, 0.05, 0.05, 0.05],
)
This example defines a complete biallelic library of length L=3, with alphabet {A, B} at every position.
Note
The mutations dict is required. epistasis-v2 uses it to build the encoding table that maps each sequence position and letter to a numbered mutation index. If a position has only one allowed letter it contributes no mutation index and is treated as invariant.
Key attributes¶
Once constructed, a GenotypePhenotypeMap exposes several attributes that epistasis-v2 reads internally:
binary_packed: a uint8 array of shape (n_genotypes, n_bits). Each row encodes one genotype as a binary vector: 0 for the wildtype letter at a position, 1 for the mutant letter. The epistasis design-matrix kernels consume this array directly.
encoding_table: a pandas DataFrame with one row per (position, letter) combination. It records the site_index, mutation_index, wildtype and mutation letters, and the site_label used for human-readable coefficient names. encoding_to_sites reads mutation_index from this table to build the list of interaction sites.
# Inspect the encoding table
print(gpm.encoding_table)
# Inspect the binary packed representation
print(gpm.binary_packed)
# array([[0, 0, 0],
# [0, 0, 1],
# [0, 1, 0],
# ...], dtype=uint8)
Tip
You do not need to interact with binary_packed or encoding_table directly in normal use. Pass the GPM to a model via model.add_gpm(gpm) and the library builds everything from there.
Attaching a GPM to a model¶
Once you have a GPM, attach it to any epistasis model with add_gpm:
from epistasis.models.linear import EpistasisLinearRegression
model = EpistasisLinearRegression(order=2, model_type="global")
model.add_gpm(gpm)
model.fit()
print(model.epistasis.values)
add_gpm calls encoding_to_sites to enumerate every interaction site up to order, builds the EpistasisMap, and caches the site list so that fit and predict can assemble the design matrix on demand.