Discriminant analysis: EpistasisLDA and EpistasisQDA¶
EpistasisLDA and EpistasisQDA classify genotypes as viable or nonviable using
linear and quadratic discriminant analysis. Like EpistasisLogisticRegression,
they binarize the phenotype at a threshold and fit over the additive-projected
design matrix, so they share the same add_gpm -> fit -> predict workflow as
every other epistasis model.

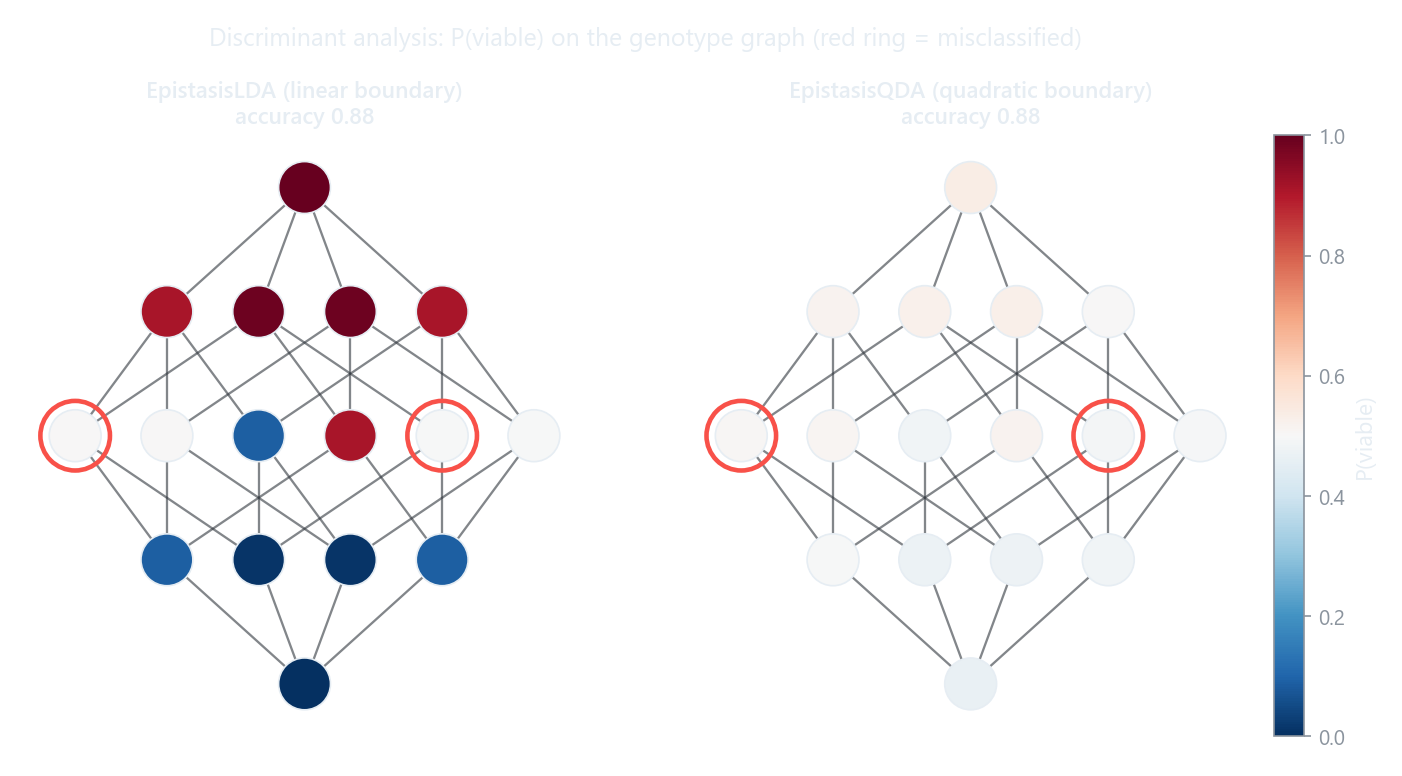
The two differ in the shape of the decision boundary they can draw. LDA assumes a
single shared covariance across both classes and produces a linear boundary;
QDA fits a separate covariance per class and produces a quadratic one. QDA is
the more flexible model but estimates many more parameters, so on small libraries
it is less confident (its probabilities sit closer to 0.5) and usually wants
regularization via reg_param.
When to use which¶
- LDA when the two classes are roughly linearly separable in the projected feature space, or when data is scarce. Fewer parameters, more stable.
- QDA when you expect the viable and nonviable classes to have genuinely
different spreads and a curved boundary, and you have enough genotypes to
estimate per-class covariances (regularize with
reg_paramotherwise).
If you only need a linear boundary with calibrated probabilities,
EpistasisLogisticRegression is the simpler choice.
How it works¶
Both models reuse the classifier base procedure:
- An order-1
EpistasisLinearRegression(model.additive) is fit to the continuous phenotypes to learn each mutation's additive contribution. - The design-matrix columns are scaled by those additive coefficients, projecting them onto a per-mutation contribution scale.
- sklearn's
LinearDiscriminantAnalysis/QuadraticDiscriminantAnalysisis fit on the projected matrix using binarized labels (y > threshold -> 1).
Constructor parameters¶
EpistasisLDA¶
threshold(float, required)- Phenotype cut-off. Genotypes with phenotype strictly greater than
thresholdare class 1 (viable). model_type(str, default"global")- Design-matrix encoding:
"global"(Hadamard) or"local"(biochemical). solver(str, default"svd")- sklearn LDA solver.
"svd"handles collinearity without a covariance inverse; use"lsqr"or"eigen"if you wantshrinkage. shrinkage(str | float | None, defaultNone)- Covariance shrinkage (only with the
"lsqr"/"eigen"solvers)."auto"uses the Ledoit-Wolf estimate. priors(np.ndarray | None, defaultNone)- Optional class priors.
EpistasisQDA¶
threshold,model_type,priors- As above.
reg_param(float, default0.0)- Regularizes each per-class covariance toward the average eigenvalue. Raise it
(for example
0.2) when the projected feature count approaches or exceeds the number of genotypes, which otherwise makes a class covariance singular.
Workflow¶
import numpy as np
from epistasis.models.classifiers import EpistasisLDA, EpistasisQDA
threshold = float(np.median(gpm.phenotypes))
lda = EpistasisLDA(threshold=threshold)
lda.add_gpm(gpm)
lda.fit()
qda = EpistasisQDA(threshold=threshold, reg_param=0.2)
qda.add_gpm(gpm)
qda.fit()
labels = lda.predict() # 0 / 1 class labels
p_viable = qda.predict_proba()[:, 1] # P(viable) per genotype
accuracy = lda.score() # fraction correctly classified
Key methods¶
| Method | Returns | Description |
|---|---|---|
fit(X=None, y=None) |
self |
Fit the additive model then the discriminant classifier. |
predict(X=None) |
np.ndarray[int] |
Predicted class labels (0 or 1). |
predict_proba(X=None) |
np.ndarray[float] |
Class probabilities, shape (n_genotypes, 2). |
score(X=None, y=None) |
float |
Classification accuracy. |
hypothesis(X=None, thetas=None) |
np.ndarray[float] |
Probability of class 1 for each row of X. |
See also EpistasisLogisticRegression,
EpistasisGaussianProcess, and
EpistasisGaussianMixture.